非综合症的先天性低促性腺激素性腺功能减退症:临床表现和基因型-表型关系(一)


(全文较长,约1.5万字,将分两篇连载,这是本文的第一篇)
Fre´de´ric Brioude1,2,3,4, Je´roˆme Bouligand1,3,4, Se´verine Trabado1,3,4, Bruno Francou1,3,4, Sylvie Salenave2,4, Peter Kamenicky1,2,4, Sylvie Brailly-Tabard1,3,4, Philippe Chanson1,2,4, Anne Guiochon-Mantel1,3,4 and Jacques Young1,2,4
1Universite´ Paris-Sud 11 and INSERM U 693, Faculte´ de Me´decine Paris Sud, F-94276 Le Kremlin Biceˆtre, France, 2Service d’Endocrinologie et des Maladies de la Reproduction, Centre Hospitalier Universitaire de Biceˆtre, 78 rue du Ge´ne´ral Leclerc, F-94275 Le Kremlin Biceˆtre, France, 3Service de Ge´ne´tique Mole´culaire, Pharmacoge´ne´tique et Hormonologie, Le Kremlin Biceˆtre, France and 4Centre Hospitalier Universitaire de Biceˆtre, Assistance Publique Hoˆpitaux de Paris, F-94275 Le Kremlin Biceˆtre, France
摘要
先天性低促性腺激素性腺功能减退(Congenital hypogonadotropic hypogonadism, CHH)起因于异常促性腺激素分泌,以损害青春期发育为特征。CHH由GNRH释放缺陷或是垂体内促性腺物质细胞功能障碍所致。与CHH有关的遗传异常的鉴别提供了生殖轴发育、成熟和功能关键路径的重要观点。Kallmann综合症中,CHH与GNRH神经元个体发生以及嗅觉缺失或减退有关,已经发现了Kallmann综合症的五种基因突变。
在多种垂体激素缺乏或复杂综合病症的CHH中,促性腺激素缺乏是伴随的、或是更复杂的内分泌疾病或非内分泌疾病的一个方面,已有研究报告了影响GNRH和/或促性腺激素分泌的其它突变。
通常CHH表型与单纯促性腺激素分泌缺乏紧密联系。这些病人没有不依赖于促性腺激素和性类固醇缺乏的相关征兆和激素缺乏,而仅是单纯CHH。在某些家庭病例,是由于遗传改变,影响GNRH分泌(GNRH1, GPR54/KISS1R,TAC3和TACR3突变),或影响促性腺激素细胞(GNRHR)的GNRH敏感性。少数Kallmann综合症或综合症形式的CHH病人也看来像是单纯CHH,但是紧密联系临床的、家庭的和遗传学研究可以再确定这种疾病的诊断,对于辅助生殖医学的遗传咨询具有重要作用。
本综述针对发表的单纯CHH病例,报告临床的和内分泌特征、遗传学病因和基因型-表型的关系。
European Journal of Endocrinology. 2010, 162: 835–851.
前言
先天性低促性腺激素性腺功能减退(Congenital hypogonadotropic hypogonadism,CHH),某些作者也称为特发性低促性腺激素性腺功能减退,是男女孩青春期障碍的病因,通常是由于两种垂体促性腺激素-LH和FSH分泌不足,阻碍了促性腺激素轴生理激活期正常的睾丸/卵巢内分泌与外分泌的功能。根据民间和军事医院病人系列,CHH的发病率男在1/4000至1/10000,是女性发病率的2-5倍(表1),但对女性可能低估了发病率。单纯的或外表单纯形式的CHH(病人本身不能自身发现嗅觉缺失或减退的Kallmann综合症)通常在青春期或成年期因青春期发育不完全或缺乏才被发现,这样的病人占内分泌医生诊治病人的大部分。

有更复杂综合症状的病人很少由成年内分泌医生发现,而常常是在儿童期因为性腺外表现由儿科医生诊断,例如GH缺乏或多发性垂体激素缺乏引起的身高生长延迟、肾上腺障碍、或肥胖、以及神经学疾病,这些疾病在青春期前出现并表现明显。
近20年来,根据遗传学和病理生理学特征逐渐对CHH进行了分类。总体来讲,现在CHH被分类为继发于促性腺激素轴和特定GNRH神经元发育异常(个体发生)的不同形式。其范例为Kallmann综合症,一般同时出现CHH和嗅觉缺失,最近对这种综合症进行了全面的综述。
最多的CHH亚组病人的表型(包括隐睾症)可完全由出生前后促性腺激素和性类固醇缺乏来解释,几乎与Kallmann综合症或其它综合症所观察到的嗅觉疾病无关。这些CHH病例继发于单纯的神经内分泌信号转导级联功能性异常,不存在已知的下丘脑-垂体-促性腺激素轴发育性或解剖性的疾病。
无论其病因和机制如何,也根据对脉冲式GNRH处理的反应对CHH分类。这种方法将下丘脑GNRH分泌异常引起的下丘脑性促性腺激素缺乏与GNRH反应异常引起的垂体促性腺激素缺乏区分开来。
几年前,根据异常自发性LH分泌的性质提出了神经内分泌分类。本文中,将无脉冲式LH分泌的的病人与LH脉冲频率或幅度减小的病人,以及只在夜间LH分泌的病人加以区别。因为发现了几种遗传性缺陷,以及对CHH病理生理学机制了解的进展,在一种综合症形式的病人可见特定的LH分泌特征,例如Kallmann综合症或有正常嗅觉的单纯CHH。由于不同的机制,LH分泌特征缺乏CHH分类的特异性。此外,费用昂贵和费时的方法的诊断意义也不大,目前仅用于临床研究的目的。
临床表现
在CHH男子,包括最严重受累的病人,不存在性别的不确定,虽然有时表现出生长不足,但这些病人都有典型的外生殖器官。泌尿生殖窦的男性化依赖于胎盘人绒毛膜促性腺激素(human chorionic gonadotropin,hCG)刺激胎儿间质细胞分泌睾酮,hCG是这些病人妊娠期间正常分泌的垂体外促性腺激素。
男孩很少在青春期前被诊断出CHH,而是通常根据新生儿期的单侧或双侧隐睾诊断。这些病例可以6月龄前的激素测定来证实,因为这时期是唯一的睾酮和促性腺激素不足的儿童期。如果确定了6月龄后“生理性”促性腺激素缺乏,根据隐睾症和/小阴茎只能得出疑似诊断,因有时与特定病因提示症状有关。例如,对于有嗅觉缺失和镜像运动的青春期前病人,可能被怀疑为Kallmann综合症,特别是在有阳性家族病史的情况下。后者可经遗传学分析证实,当家族已经鉴别出突变时,这种分析是很容易的。但这种综合症,特别是常染色体形式的综合症,必须谨慎预测可能的成年期促性腺激素表型,因为,甚至在同一家族中,一定突变存在表型的可变性。
在大部分病例,在女13岁和男14岁后,青春期发育不完全或不出现,那么将存在有CHH的可能性。如果青春期延迟同时有隐睾症或小阴茎,或是在18岁以后继续存在,那么这种诊断的可能性就很大。当在年龄较大而诊断为CHH时,因肾上腺正常分泌的雄性激素在外周转换成睾酮,病人可能已有阴毛,可能使无经验的医生认为是青春期开始,而身体检查表现出青春期前睾丸体积或至少为严重生长不足的睾丸。
根据生长形式,通常可将单纯CHH的病人与体质性青春期晚出现病人相区分,因为前者有正常的年龄身高,而后者倾向于较矮。不同综合症状表现对于这两种情况的区分很有帮助,否则存在困难。对于有隐睾症和/小阴茎的严重CHH病人容易诊断,而对部分或中间形式的病人,仅根据临床更加难以与体质性青春期晚出现区分开。
在未治疗的CHH病人可见男子乳腺发育,但是,通常发生在以hCG或超生理剂量睾酮治疗时。在这种情况下,睾酮的芳香化可能导致睾酮∶雌二醇比例减小。因为肾上腺雄性激素分泌与促性腺激素轴无关,因而单纯性CHH病人肾上腺雄性激素分泌正常,所以肾上腺雄性激素也导致男子乳腺发育在理论上也是可能的。
似乎仅少数病人受累于不完全性先天性促性腺激素缺乏,以一定程度的男性化和男子乳腺发育为特征,而且睾丸体积在4ml以上或甚至接近正常。但是,随分子遗传学的发展,家族性遗传研究和亲属测序已经能够诊断出未自发就医的很不完全形式的病人,以及有中等临床和内分泌异常的病人。
在儿童期身高生长正常的所有CHH病人,尽管无青春期生长突增,身高生长延迟也很少是表现症状。相反地,长骨骺不闭合解释了这些病人时常出现的类无睾外表和相对较高的身高。当在成年期发现促性腺激素缺乏时,也常见骨成熟延迟、骨量减少和骨质疏松。
在约30年前,在几例病人确定了所谓的可逆形式,主要是在男性Kallmann综合症病人。开始时根据临床确定, Quinton et al.在1999年和最近的Raivio et al.根据UK病人系列,详细描述了其临床和激素特点。在KAL1, FGFR1, GNRH 受体基因 (GNRHR) , 以及PROKR2突变病人证实了它的存在。可逆形式与脉冲式促性腺激素很晚激活有关,由于GNRH脉冲发生器和/或促性腺激素细胞激活晚,促性腺激素分泌随时间进展而得到改善。如果未进行内分泌治疗或睾酮处理,睾丸体积增加,那么就可能是这种临床变异体。
在嗅觉正常并无可鉴别突变的病人,20岁以前出现这些可逆形式时,将难以和体质性青春期晚出现者相区别。
90%以上的女性CHH病例是通过原发性闭经所发现的。乳房发育可能高度可变;通常出现乳房发育,有时几乎正常。同样,阴毛可能缺如、稀少或甚至正常。在女性常见的这些不完全形式可能解释了低估的女性发病率。迄今,仅在少数女性报告了很轻微表现形式的病例,因单纯的持续不排卵而发现CHH,然而雌二醇分泌与子宫内膜发育相一致,表现为孕酮处理后开始出血,以及存在月经或月经稀发。在自然怀孕女性也已经报告了这些减轻症状的疾病表现。
对于嗅觉正常并无可鉴别突变的散发CHH女性,其难点是与功能失调性下丘脑性闭经的鉴别诊断。对于有提示HH的激素特征的,但无嗅觉缺失或减退或可鉴别的遗传异常的原发性闭经女性,在排除低体重,饮食失调、过度活动和慢性基础疾病后,必须谨慎考虑CHH,必须跟踪随访这些病人,根据其身体组成重复估价促性腺激素轴和排卵机制。当体重或体重指数(BMI)在正常值下限时,也可以使用身体脂肪指标筛查功能失调性下丘脑性闭经。
激素测定调查
在男性,根据低血浆总睾酮水平、同时LH和FSH水平下降来诊断低促性腺激素性性功能减退。完全性促性腺激素缺乏病人很容易诊断,大部分病人的总睾酮水平低于1ng/ml(3.467nmol/l)或0.5ng/ml,促性腺激素缺乏病人的另外一种睾丸激素-抑制素B也是低的。在CHH,这就说明FSH缺乏所决定的塞尔托利细胞(Sertolian cell)功能不足。但是,抑制素B并非是低促性腺激素阳性诊断的充分敏感性指标,因为在不完全性病人可能是正常的。然而,当以睾丸体积修正时,抑制素B可为促性腺激素损害程度提供有价值的信息。
循环抗-缪勒激素浓度保持在青春期前水平是男性CHH的另一个特征,反应了缺乏睾丸内睾酮引起的青春期睾丸成熟。但是,因这种塞尔托利细胞激素的复杂调节-与睾丸睾酮和FSH的调节方向相反,如用于诊断需非常精细的了解,尚待进一步的估价。
虽然GNRH试验在广泛使用,但因成本效益一般,对其实用价值产生了许多的异议。的确,GNRH试验未能比当代测定方法提供更多的关于基础促性腺激素水平的诊断信息。此外,也不能说明下丘脑或垂体源的促性腺激素缺乏:在CHH病人和青春期后HH病人,对于完全性下丘脑性促性腺激素缺乏可能是阴性结果,而对不完全垂体性缺乏则为阳性。先天性促性腺激素缺乏病人对GNRH试验的反应高度可变,并通常依赖于男性睾丸萎缩和女性乳房发育所反映的缺乏严重程度。因此,完全缺乏并睾丸体积在2ml以下的男性,反应通常很微弱或无反应,而部分缺乏并睾丸体积在6ml以上的男性反应可能为阳性,甚至有超常的LH。总之,GNRH试验更多的用来证实临床上已经发现的先天性促性腺激素缺乏的严重程度,而不用于阳性诊断。
CHH女性病人的血浆雌二醇浓度通常较低,有时接近检测下限。雌二醇浓度似乎与乳房发育相关:在乳房无发育者,循环雌二醇浓度很低(通常不可检测),而当乳房发育超过B2期时,使用敏感的测定方法可以检测到雌二醇。青春期发育与促性腺激素浓度之间存在类似的关系:在乳房无发育时促性腺激素浓度很低或不可检测,在乳房B3或B4发育期时,促性腺激素浓度可接近正常月经女性初期卵泡时相的水平。
在做出可靠的单纯先天性促性腺激素缺乏诊断之前,必须检查垂体前叶的功能,以免遗漏高催乳素血症、全部垂体前叶功能不全,或可能与部分CHH综合症形式有关的内分泌疾病。
在无隐睾症和嗅觉缺失情况下,原发性青少年血色素沉积症可能与CHH相似。因此,应排除铁过多,测量血清铁和转铁蛋白饱和系数可排除原发性青少年血色素沉积症。
脑和嗅球磁共振成像(MRI)有益于CHH的诊断。虽然特发性嗅觉正常CHH的MRI总是正常的,但MRI可以排除下丘脑-垂体区域的膨大、侵润或畸形疾病。在搜索卡尔曼综合症症状中,MRI也可用来分析嗅球和裂沟(单侧或双侧发育不全,以及裂沟的消退),例如,当不能进行半量的嗅觉测量时。MRI也可用于检查其它解剖学结构,例如,可能与卡尔曼综合症或Gordon Holmes综合症有关的胼胝体和小脑。
卡尔曼综合症的肾脏超声检查有特别的意义,因为可发现肾脏的畸形或发育不全,特别是在KAL1异常的病人,而单纯CHH病人则总是正常的。睾丸和内生殖器官的超声检查对于准确确定睾丸体积,评价精囊和输精管有很大的价值,如果存在异常,可能需要进行不育治疗。
在女性骨盆超声可以确定与促性腺激素缺乏严重程度有关的子宫大小、以及子宫内膜厚度、卵巢大小和卵泡数量和大小。
单纯性非综合症的CHH
在确定病人为这类疾病之前,必须先进行细致的身体检查和家族史调查,以排除轻微的综合症形式。病情检查必须全面,并包括嗅觉半定量估价(嗅觉测量法),因与嗅觉完全缺失不同,简单的访谈难以检测出嗅觉减退。也应当询问病人的家庭,如果可能,可鉴别出以前未发现的嗅觉缺陷。这样做是重要的,因为通过广泛使用系统的调查方法和受累家庭的突变筛查,可鉴别出开始认为无症状的或貌似单纯CHH的,但实际为不完全或完全嗅觉缺失的携带突变的亲属。仔细检查外耳和听觉也是重要的,以排除可能被误认为非综合症CHH或卡尔曼综合症的轻微CHARGE综合症。
垂体GNRH抵抗和GNRH基因功能丧失突变引起的CHH
GNRHR基因缺陷是最早鉴别出的非综合症CHH的病因。我们实验室中对单纯CHH病人编码GNRHR基因的系统分析,使我们在1997年发现引起一名男性及其非同宗姐妹GNRHR功能丧失的突变,二者为有正常嗅觉的不完全性CHH。虽然病人及其另外一名姐妹是杂合子并有正常的表型,但这两名病人为复合杂合子,说明是隐性常染色体遗传疾病。位于细胞外第一环路的Glu106Arg突变部分抑制了GNRH与其受体的结合。相反,位于第三胞内环路C端部分的Arg262Glu突变并未改变GNRH的亲和力,而是影响了信号转导。
自首次报告以来,在完全和不完全促性腺激素缺乏男女受试者,报告了许多家族性和散发性促性腺激素缺乏和GMRHR突变病例,图1。
所鉴别出的相对大量病人可以确立GNRHR信号转导抑制程度与不完全或完全性CHH之间,以及与对外源性GNRH脉动处理的急性或长期阳性、阴性反应之间的基因型-表型关系。但是,在无关受试者,甚至在有相同突变和不同程度促性腺激素缺乏的家庭,体外受体功能研究结果与临床表型之间存在冲突。当表型严重和非脉冲式LH分泌时,GNRHR突变病人最一致的特征是对GNRH脉冲处理的垂体抵抗。这种激素特征可将这些病人与GPR54/KISS1R, TAC3, 或 TACR3突变的病人区分开来,因为后者病人,甚至在严重程度下,也对外源性GNRH有高度敏感性,就如同我们将下面所讨论的那样。在GNRHR突变的和不完全促性腺激素缺乏的CHH病人,可能自然脉冲式释放LH,但脉冲幅度表现可能低于正常。类似地,高剂量GNRH可能引起不完全性病人的反应,并且LH脉冲幅度增加。有这样的一个病例,脉动式GNRH处理后获得了妊娠。在我们最初报告的这名女性病例中,在克罗米芬处理后,两次排卵并成功相继妊娠。这种反应提出了病人促性腺激素细胞受到由克罗米芬引起的内源性GNRH分泌增加的可能性,尽管她有部分受体功能障碍。
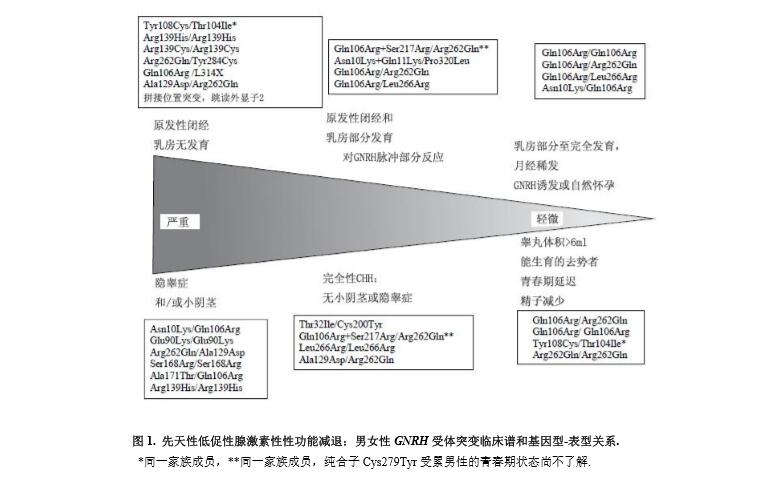
GNRHR突变似乎是家族性嗅觉正常CHH最常见的病因,在某些病人系列中出现率接近40%,很少发现散发病例(5%以下)。
在大部分早期报告中,GNRHR缺陷包括引起氨基酸置换的点突变而导致受体功能障碍,特异影响配体结合或信号转导,未改变GNRH与受体的结合。引起移码或提前终止密码子的突变很少见,但至今为止尚无真正GNRHR1缺失的报告。
GNRHR1点突变分布于全部受体编码序列,包括NH2终端、跨膜区域2-7、细胞外环1和2、细胞内环3。已经鉴别出两个突变热点-Gln106Arg 和 Arg262Gln突变。在不完全性和完全性CHH病人,已经报告了GNRHR的六种纯合子和十种复合杂合子突变。在异种细胞系统的17种GNRHR突变的体外表达证明,某些突变是完全非功能性的(Glu90Lys, Ala129Asp, Arg139His, Ser168Arg, Ala171Thr, Cys200Tyr, Ser217Arg, Leu266Arg, Cys279Tyr, Pro320Leu, 和 Leu314Xstop),而其它突变的功能程度可变(Asn10Lys, Asn10Lys_Gln11Lys, Thr32Ile, Gln106Arg, Arg262Gln, and Tyr284Cys)。
关于GNRHR1功能的损害,虽然早期在体表达研究提出了突变仅改变了分子的特异功能(配体结合、受体激活、或与耦合效应器相互作用),但Michael Conn实验室的一些研究结果对这些简单化的观点提出了挑战,指出了GNRHR蛋白质错误折叠和因此的错误指向是导致人类GNRHR(hGNRHR)功能丧失的机制。在令人关注的综述中,这个研究组总结了导致这个结论的实验数据,解释了这些研究是如何揭示了以前未被怀疑的hGHRHR突变。此外,他们也提供了挑战传统基因型-表型关系研究关于突变体hGNRHR结构分别表达的数据,提示一定复合杂合子表型与功能丧失程度主要由较少严重受累的等位基因所确定。他们证明了这种假设未考虑到一定hGNRHR突变体对其它人的潜在显性负效应。