非综合症的先天性低促性腺激素性腺功能减退症:临床表现和基因型-表型关系(二)


(本文较长,约1.5万字,分两部分连载,这是本文的第二篇)
携带GPR54/KISS1R突变病人的CHH
在2003年,两个研究组分别在家族性和散发性嗅觉正常CHH病人鉴别出了GPR54/KISS1R功能丧失突变,也发现了这些受体及其假设配体- KiSS1基因编码的吻肽(kisspeptins)的生殖作用。这一发现是当代神经内分泌学的重大突破,将下丘脑GNRH和垂体促性腺激素分泌生理学控制的研究集中到该受体及其配体的作用上来。在最近的许多综述中都谈到了这些问题,因此我们将集中讨论这些突变病人的临床与内分泌特征,特别是共同的和/或特异特征。
2003年九月,de Roux et al.发表了嗅觉正常CHH的GPR54/KISS1R突变的第一篇报告。病人为法国8名儿童的家庭成员,5名有CHH。索引病例为因青春期延迟而转诊的20岁男性,具有典型的性腺功能减退症,有小阴囊内睾丸(4ml)、阴毛稀疏、阴茎7cm、骨龄15岁,嗅觉正常并无相关的异常,三名兄弟有类似临床特征,但在第一篇报告中未提供细节。他的16岁的妹妹有不完全性性腺激素功能减退,乳房部分发育,曾有一段时间的子宫出血。激素评价表明,所有受累男性有低水平的血浆睾酮,所有受累女性有低水平的血浆雌二醇,所有病人都有低血浆促性腺激素水平,对GNRH刺激试验有不敏感至“正常”的促性腺激素反应。受累同胞的测序分析表明,在跨内含子4至外显子5拼接受位点和外显子5的部分,存在155bp的纯合性缺失。在非受累家庭成员中,有无缺失仅发生在一条等位基因上,指明为常染色体隐性遗传。
仅在一个月后发表的第二篇文献中,Seminara et al.报告了一个有血缘关系的沙特阿拉伯大家庭,有3名为第一级堂兄妹间结婚,有6名受累和13名未受累后代。所有受累的个体都携带有外显子3的纯合性单核苷酸突变(443T>C),在148位置上的亮氨酸被丝氨酸替换(Leu148Ser),导致体外的GPR54功能丧失。最早由Bo-Abbas以及后来的Pallais,已经报告了该家系的临床和激素特征。受累个体的临床和内分泌学评价揭示,有低水平的性类固醇与促性腺激素水平,证实了CHH诊断。后来某些病人进行了生育力治疗。有趣的是,接受外源性促性腺激素的4名受累男性睾丸发育成熟,精液中出现精子,随后具有生育力,大体上排除了原发性睾丸功能障碍。同样,一名接受脉冲式GNRH治疗的女性,引起排卵和受孕。
同一研究组所发表的报告详细描述了另外一名GPR54/KISSR的Arg331X和X399Arg杂合性功能丧失突变的男性病人。这名病人因青春期延迟而在17 10/12岁被发现,具有典型的完全性腺功能减退的身体症状(面部无毛、稀疏的腋毛和阴毛、睾丸生长不足(1.2ml)、阴囊内睾丸、非小阴茎),嗅觉正常。因此,血浆睾酮水平很低,促性腺激素水平也很低,但对GNRH刺激的反应显著。睾丸或组织检查表明为精子缺乏,初期发育不全和早期萎缩。该名病人对外源性脉冲式GNRH的敏感性显著高于所有进行类似治疗的病人,在长期治疗中睾丸体积增加并精子生成,在治疗1年和1年半后睾丸体积达到12ml时,他的妻子怀孕,后来的精子分析表明精子数为7百万/ml。
在Lanfranco et al.的文章中,KISS1R 突变的渊源者有CHH和双侧隐睾症,是德国一家庭中的唯一的孩子。在31岁时想要孩子而开始GNRH治疗,刺激精子生成。几个月后测定的睾酮水平正常,而促性腺激素水平显著增加(LH: 11.5IU/L;FSH:24.9IU/L),说明了可能是由于隐睾症而睾丸损害。同时,超声扫描发现两侧生长不足的阴囊内睾丸(右侧:3ml;左侧:5.3ml)。在经2年脉冲式GNRH治疗后的33岁时,精子分析表明为永久性少弱精子症,但尽管如此,经过IVF他成为一名健康儿子的父亲。GPR54/KISS1R测序证明,在核苷酸1001位置后胞嘧啶纯合性插入,导致开放读框中的移码,由氨基酸301开始,包括了第七跨膜区域,因此由398至441氨基酸拉长了GPR54蛋白。这种突变的功能性后果虽然可能有害,但尚未在体外检验。
Semple et al.报告了一名正常妊娠足月出生的土耳其-塞浦路斯与加勒比黑人混血男性渊源者。该名男子有小阴茎,出生时未下降到阴囊的睾丸,在2个月时,时机窗口的血清促性腺激素几乎不可检测(LH:0.5mIU/ml,FSH:0.5mIU/ml),垂体其它功能正常。发现其为GPR54两种错义突变的复合杂合子,即第5跨膜区域的半胱氨酸297至精氨酸(C223R)、第3细胞外区域环中的精氨酸297至亮氨酸(R297L)。在细胞稳定表达GPR54中,发现C223R突变使信号发放受到极大的损害,而R297L变异体信号发放仅有轻微的损害。
最后,Tenenbaum-Rakover et al.在叙利亚和以色列的两个非血缘关系的阿拉伯穆斯林家庭中,鉴别出了5名CHH病人。所有受累受试者为另外一种GPR54/KISS1R错义突变纯合子,即残基102 (L102P)的亮氨酸被脯氨酸替代,使GPR54信号发放完全消失。该病人因原发性闭经和部分青春期发育(Tanner B4)而就诊,治疗前的下丘脑-垂体-性腺轴评价证实了HH,骨盆超声揭示小子宫和小卵巢,直径<10mm的卵泡不足5个。重要的是,后来以外源性脉冲GNRH治疗,引起排卵和两次正常的妊娠。
在第二个家庭中,渊源者出生于非同源的父母。出生时发现小阴茎和隐睾症。他有11个兄弟姐妹,包括2名原发性闭经,但乳房部分发育(Tanner B3)、阴毛发育良好,并有典型CHH激素特征的16和17岁的姐妹。在第二个家庭中,有不同程度的性腺功能减退,GNRH试验也表明LH和FSH反应也具有家庭内的可变性,男性渊源者的LH反应迟钝,而姐妹LH增加显著。该文作者报告,渊源者的激素研究揭示了正常频率的持久性脉冲式LH分泌,但振幅很低,提示了GPR54是失活性损害,但未妨碍青春期内分泌的开始。
虽然至今报告了GPR54/KISS1R突变病人的数量相对少(不足20例),但某些共有的临床和内分泌学特征应引起注意。
与持有GNRHR突变病人一样,携带GPR54/KISS1R突变的病人都有嗅觉缺失或其它在Kallmann综合症或其它综合症形式的CHH所见到的相关症状。
当以GNRH或促性腺激素治疗时,所有KISS1R突变病人都出现反应,有力地说明了GPR54/KISS1R功能丧失突变并未降低促性腺激素细胞对GNRH的敏感性,或者未降低性腺对促性腺激素的敏感性。因此,如同在鼠一样,人类GPR54/KISS1R功能丧失似乎主要影响下丘脑GNRH的分泌,对于垂体或性腺无可辨别的直接影响。所以,在gpr54?/?小鼠和野生小鼠所观察到的类似下丘脑GNRH浓度说明了这些神经元在发育中迁移正常,保持有GNRH的十肽生物合成能力。因而,GPR54/KISS1R系统似乎主要参与下丘脑GNRH的释放。这一点得到大鼠和羊功能研究直接证据的支持,在这些研究中证明了吻肽可刺激下丘脑GNRH的分泌:吻肽-10引起大鼠下丘脑外植体的GNRH释放;在羊,大脑内吻肽注射引起GNRH释放入脑脊液。这些数据都有力提示,吻肽控制促性腺激素轴的基本位点在下丘脑GNRH神经元。对这种观点的进一步的支持来自于大鼠的表达和功能研究,77%的GNRH神经元共表达GPR54/KISS1R mRNA,吻肽有效地引起>85%的GNRH神经元c-fos(激活的早期标志)表达。此外,原位电生理学记录证实了吻肽刺激GNRH神经元的能力,即吻肽能够激起成年小鼠下丘脑>90%GNRH神经元群长时间的去极化反应。总之,来自不同动物模型的重要证据证实了吻肽能够直接激活下丘脑GNRH神经元,这是对促性腺激素释放有强力诱导作用的主要机制基础。
鉴于上述总结的发表数据,GPR54/KISS1R突变病人对GNRH表现出显著的垂体敏感性,这种十肽的脉冲式处理是合理和有效的,即使是使用较低剂量和在严重CHH的受试者。这种响应性与GNRHR突变病人形成鲜明对照,GNRHR突变病人除了轻度者外,抵抗这种十肽。
在CHH和GPR54/KISS1R突变男性,另一有意义的发现是有隐睾症和小阴茎病人的比例相对高。隐睾症反映了出生前促性腺激素缺乏,因此在青春期前很久这些病人也似乎存在促性腺激素缺乏。Semple et al.的发现更加强调了这一点,在新生儿期,他对这样一名病人的评价证实了促性腺激素缺乏的存在。这些数据表明,并不像最初所说的那样,下丘脑Kiss/GPR54信号发放的激活不是启动青春期所特定必需的,而是它参与促性腺激素轴生理性激活的所有阶段。
GPR54/KISS1R纯合子或复合杂合子突变的所有个体都有青春期发育障碍,杂合子家庭成员有正常的生殖表型,说明单基因隐性遗传与GNRHR突变一样,在家族渊源者比非家族渊源者中似乎更多见KISS1R突变。但是,在GNRHR所证实的突变比在KISS1R更普遍,必须对GPR54/KISS1R突变病人进一步的研究,以确定后者基因突变所导致的表型谱,也必须估价嗅觉正常的非综合症CHH病人群的准确发生率。
神经内分泌控制促性腺激素功能的两种新作用物—TAC3和TACR3编码的神经激肽B及其受体NK3R
2009年12月看到了一篇重要报告,描述了意外影响家族嗅觉正常CHH两条基因的突变。Topaloglu et al.通过对土耳其9名先天性CHH的家庭基因组单核苷酸多态性(SNP)分析,确定了神经激肽B在人类生殖轴中的的重要作用。这些土耳其家庭中严重先天性促性腺激素缺乏的男女性存在TAC3纯合子错义突变,导致一种速激肽家族神经肽-神经激肽B,或TACR3编码的7个跨膜域受体NK3R的功能性丧失。
在其中的3个家庭,作者在NKR3的跨膜区域发现了纯合突变(编码Gly93Asp和Pro353Ser),并在一个家庭发现位于成熟神经激肽B的速激肽基序中的Met90Thr置换。
家谱分析也揭示Met90Thr置换的杂合子携带者未受累,说明了常染色体隐性CHH,因此,无单倍体不足或突变的显性负效应。功能分析证实,当在异种细胞系表达时,配体和受体突变导致体外受体信号发放受损害。 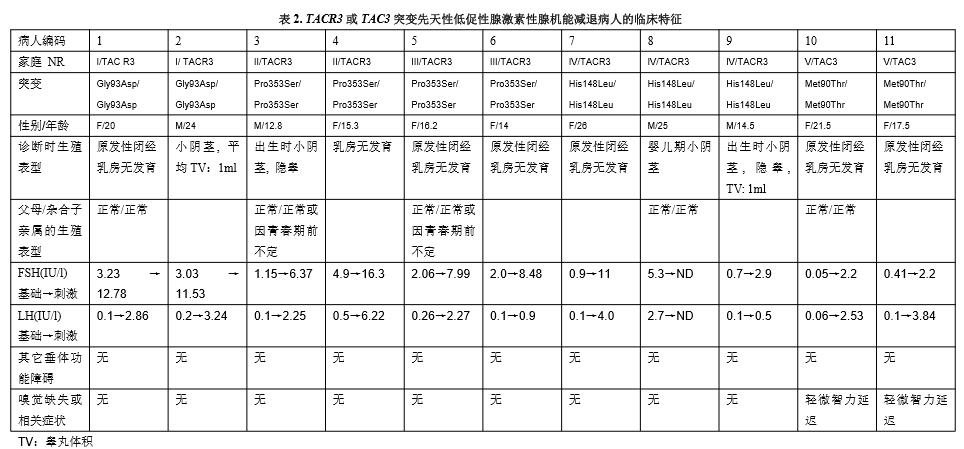
在最近的一篇文献中,同一研究组提供了神经激肽B及其受体调节人类生殖功能的进一步证据。他们在有3名nCHH兄弟姐妹的家庭对TACR3基因测序,发现所有受累同胞为NKB受体第一细胞外环His148Leu突变的纯合子。当使用钙离子流作为功能读出时,His148Leu突变受体对NKB表现出明显的信号发放损害。
神经激肽B和NK3R影响生殖内分泌控制的确切机制尚待确立。作者提示,可能是影响下丘脑GNRH的释放,表达GNRH的啮齿动物神经元表达NK3R。此外,在解剖学上,下丘脑正中隆起内表达神经激肽B的神经元轴突与GNRH神经元并列,NKB-免疫反应性膨体与GNRH免疫反应性轴突直接接触。最后,在弓状核中含有雌性激素受体-α和强啡肽23的区域,NKB的表达量最高。在对GNRH分泌的反应中,雌性激素受体-α和强啡肽23参与孕酮反馈。
世界范围内都在进行神经内分泌研究,以揭开这两种新因素影响GNRH分泌的机制。
在最近的一项研究中,我们鉴别了三名TAC3内含子3拼接接受位点(c.209-1G>C)纯合取代病人,以及三名TACR3内含子2拼接接受位点(c.738-1G>A)纯合性突变的兄弟姐妹病人。我们证明,这两种突变使神经激肽B及其受体BK3R分别无效。有趣的是,与Topaloglu et al.一样,我们在几名病人观察到很低的LH水平与正常或接近正常的FSH水平的离异,对于GNRH的刺激,促性腺激素过度反应。这种特殊的激素特征指出了神经激肽B信号发放变化病人的神经内分泌的损害。在这些病人,脉冲式GNRH处理正常化了性类固醇水平和LH的释放,在女性恢复了生育力。总之,这些结果证明,在遗传性嗅觉正常的非综合症CHH,存在下丘脑源的促性腺激素缺乏。因此,神经激肽B和NK3R对于人类下丘脑GNRH释放具有重要作用。
GNRH1突变是人类CHH的一种病因
GNRH对于哺乳动物生殖的调节至关重要,它由下丘脑神经元合成,并有神经末梢释放入门脉循环。在与膜GNRH1型受体结合后,刺激垂体前叶促性腺激素细胞合成和释放LH和FSH。然后,这两种肽进入体循环到达性腺,刺激类固醇激素的分泌并配子发生。
1977发现的缺乏GNRH鼠性腺机能减退,以及1986年证实的gnrh1缺失的作用,提示了GNRH1突变可能引起人类CHH。但在2009年才发表了GNRH1突变是CHH发病机理的证据。
在1977年,Bruce Cattanach及其同事发现了鼠自然突变体,成年雄性和雌性鼠表现出完全性性腺功能减退。他们也证明了与下丘脑GNRH缺乏并导致垂体LH和FSH减少及循环促性腺激素水平下降相关的表型(图2)。后来在hpg鼠,几个研究组提供了低促性腺激素性腺功能减退与GNRH缺乏之间的因果关系证据,他们证明,当在第三脑室放置正常胎儿视前组织(啮齿动物含有大部GNRH神经元的区域)时,使雄性和雌性hgp鼠的内分泌缺乏发生逆转。
Peter Seeburg研究组在1986年证明,hpg鼠的遗传性常染色体隐性低促性腺激素性腺机能减退是由gnrh1远侧半的33.5kb缺失所致。部分缺失的基因,缺乏编码大部分GAP的两个外显子,具有转录活性,由下丘脑组织原位杂交所证实,但是使用免疫细胞化学或使用抗GNRH前体不同部位的RIA法检测不到GNRH肽。这一研究组使用转基因技术,使hpg鼠恢复了正常的GNRH基因,观察到垂体和性腺功能完全恢复,因此而证实了gnrh1在下丘脑控制啮齿动物生殖中的关键作用。
hpg鼠的gnrh1基因分析显示了大部分的缺失,仅剩下了启动子区域,编码信号肽的前两个外显子和GAP的前11个氨基酸残基。但是,因为这种突变完整保留了编码GNRH十肽的区域,所以为什么hpg鼠性腺机能减退的原因仍然难以捉摸。一种假设是该基因的切断导致了无功能的GNRH前体mRNA分子,因此GNRH神经元不能生成和分泌GNRH。其神秘机制最终被K Kim领导的研究组通过分析GNRH前体-mRNA剪接所解释。这些作者提出,两个末尾外显子(含有外显子剪接增强子)的基因组缺失导致第一内含子转录的积聚,并被运送到细胞质,强烈抑制第一内含子转录物向GNRH肽的翻译,确定了内含子1保留对下游开放阅读框翻译活性的抑制性作用,最终导致hpg鼠功能性GNRH缺乏和性腺机能减退。
使用候选基因方法,我们花费多年来筛选非综合症嗅觉正常的CHH病人亚组,以鉴别GNRH1基因突变。在近150名渊源者的分析中,我们在2008年1月鉴别出纯合性GNRH1移码突变,这种突变在编码含有GNRH(前原-GNRH)蛋白前体信号肽的N-端区域,在核苷酸18处有一个腺嘌呤插入(c.18-19insA)。索引患者是一名年轻男性,在18岁时因青春期障碍而转诊。这名病人具有典型的完全性性腺机能减退症状,有阴囊内小睾丸、无阴毛和小阴茎,但嗅觉正常。它的一名受累的嗅觉正常的妹妹,在17岁进行评价,也为完全性性腺机能减退,乳房未发育、无月经初潮、超声检查发现小子宫和两个小卵巢。这两名病人的性腺类固醇和血清促性腺激素的水平很低,对GNRH刺激试验反应迟钝,有低至正常的促乳素水平,其它的垂体功能正常。在受累的妹妹,出现非脉冲式基线LH分泌形式,我们也有机会来研究对外源性脉冲式GNRH的垂体反应,以鉴别下丘脑或垂体缺乏。如Charlton et al.在hpg鼠所报告的那样,我们发现外源性GNRH恢复了内源性LH脉冲性,证实了下丘脑源的促性腺激素缺乏,以及垂体细胞对GNRH的正常反应性。脉冲式GNRH处理也恢复了病人的卵巢功能,雌二醇和抑制素B循环水平增加,超声图可见单一优势卵泡的募集。两名索引受试者为纯合子突变,而未受累的父母及姐妹为杂合子,具有正常的生殖表型。因此就像hpg鼠那样,这种疾病以常染色体隐性性状被遗传。这些观察提示,在人类和鼠,GNRH1和gnrh1基因每单拷贝足以使GNRH正常分泌,保持促性腺激素轴正常功能,因此排除了单倍体不足。
我们在这两名受累个体检查出的GNRH1纯合性c.18-19insA突变引起了移码,如果翻译,将引起GNRH前体氨基酸7开始的异常肽。这种异常肽的疏水核缺乏肽信号序列,完全没有正常GNRH十肽序列,有力地提示了其病理性质,总长度42个氨基酸代替了正常前原-GNRH肽。此外,我们的体外实验清楚地证明了纯合突变的有害性质:转染的GNRH1突变不能维持AtT20细胞的GNRH分泌,而在以野生型GNRH1基因转染时,相同的细胞能够产生和分泌成熟的GNRH。
值得注意的是,尽管纯合性移码突变可能导致异常肽的翻译,也缺乏GAP序列,但这两名CHH病人有正常至低水平的血浆促乳素。虽然他们长期性激素替代治疗,促乳素水平保持正常,排除了正常至低水平的促乳素仅与性类固醇激素缺乏相关的可能性,证实了生理条件下,GAP并非作用于人类垂体腺来调节促乳素的分泌,这与Seeburg研究组在1985年自然杂志上的论断不同。在这一方面,有趣的是返回到了Cattanach最初报告hpg鼠的文献上来,雄性和雌性鼠有很低的垂体促乳素含量。Nikolics的文献忽略了这一重要结果,这个结果有力地反驳了GAP在促乳素分泌中的抑制作用。
在初步证明GNRH1突变引起CHH后,又鉴别出了第二例纯合性移码GNRH1突变(G29GfsX12),可以预测GNRH十肽的3个C端氨基酸断裂,产生了过早中止密码子。后者突变在亚美尼亚的青春期前男孩所发现,该男孩因隐睾和小阴茎在8岁8个月时进行检查评价。这名病人可能为CHH,有正常的嗅觉,在13岁6个月时尚未进入青春期,但后来通过庚酸睾酮治疗而男性化。遗憾的是,无进一步的临床和激素数据来准确确定CHH诊断。有趣的是,这名男孩的父母非常可能是杂合性携带者,青春期时间正常,因为在我们所报告的家庭中,进一步地支持了CHH遗传形式的常染色体隐性性质。后来的文献也提到了在4名CHH病人鉴别出的杂合子变异体,即1名GNRH十肽第8个氨基酸非同义的错义突变、1名引起GAP过早序列终止的无义突变、2名引起非同义氨基酸置换的两名序列变异体、以及1名GAP信号肽和其它的突变。但是,杂合子GNRH1变异体对嗅觉正常CHH的发病机制的贡献仍有待于确立。
嗅觉正常的非综合症CHH、Kallmann综合症和CHH的复杂综合症病因:相互有关吗/有何相关?
如上所述,与GNRHR1, GPR54/KISS1R, GNRH1, TAC3,和TACR3突变相关的CHH表型局限于促性腺激素和性激素缺乏,这些病人没有卡尔曼综合症或是更复杂的CHH综合症的临床表现,例如CHAGRE综合症。但是,最近几年报告了一些家庭,其中某些成员患有卡尔曼综合症,而其它成员有正常或表面正常的嗅觉(因不完全是半定量的嗅觉检查法)。自从发现卡尔曼综合症5个遗传特定病因以来,已经确立卡尔曼的两个基本症状之间的分离主要见于常染色体形式的病人,而在KAL1突变的X连锁形式的病人,两种基本症状几乎总是同时存在。
一项最近的研究表明,在134名嗅觉正常的病人中,有7%的病人存在FGFR1突变,这与卡尔曼综合症病人中的这种突变发生率(10%)相接近。但是在134名中的少数病人以半定量方法评价了嗅觉,提出了某些病人为低嗅觉的可能性,意味着真正嗅觉正常的CHH病人中FGFR1突变发生率可能低于这些作者的报告的数字。
Kim et al.在197名有或无嗅觉异常的CHH病人进行了CHD7基因的突变筛查。这些作者在3名散发卡尔曼综合症病人和4名散发嗅觉正常的CHH病人,鉴别出了7种杂合突变。因而提出这些疾病可看做是CHAGRE综合症的轻微等位基因变异体。然而,如上所述,Jongmans et al.仅在有CHARGE综合症额外表型特征的卡尔曼综合症病人鉴别出了CHD7突变。因此,这项研究说明诊断为卡尔曼综合症的病人应当严格筛选CHARGE综合症的临床特征,当发现有耳聋、心脏畸形、耳畸形、和/或半规管发育不全时,应当建议进行CHD7测序。
区分真正嗅觉正常的非综合症CHH与嗅觉正常和貌似正常的卡尔曼综合症或CHARGE综合症的需要不是纯粹的理论问题,因为这也涉及到了病人处理的重要问题。的确,鉴别出突变的嗅觉正常的非综合症CHH的遗传模式似乎都是常染色体隐性。因此,在无血缘关系的情况下,可以确保嗅觉正常的非综合症CHH者辅助生殖,孩子受累的风险很低。相反,由于FGFR1突变的卡尔曼综合症,或与CHD7突变相关的CHARGE综合症常染色体显性遗传,传递风险为50%。此外,甚至在同一家庭中表型表达的可变性使得难以预测这些病人孩子的综合症严重程度或相关疾病的风险。为此,必须彻底查实貌似非综合症CHH病人的表型,包括仔细检查轻微综合症形式的指示症状。
二基因遗传
长期以来,认为嗅觉正常CHH和卡尔曼综合症是孟德尔遗传模式的单基因疾病。但是, Dode et al. 在2006年发表了第一例可能的卡尔曼综合症二基因遗传。他们报告了杂合性PROKR2的p.L173R功能丧失突变,也携带有KAL1外显子8中的p.S396L无义突变。此后,在其它卡尔曼综合症或嗅觉正常的CHH病人也证明了二基因遗传,在PROKR2 和PROK2,或在FGFR1 和 NELF 或GNRHR,以及在PROKR2 和GNRH1, GNRHR, 或KISS1R中的突变。不同基因的缺陷可能互相增效,引起CHH或卡尔曼综合症表型,或是改变GNRH缺乏的严重程度,部分解释了CHH和卡尔曼综合症家庭内和之间的表型可变性。
因此,我们必须重新考虑目前研究CHH或卡尔曼综合症遗传的过分简单的方法,不仅要考虑显性,更要考虑隐性遗传模式,因为寡基因性(oligogenicity)也是遗传咨询的重要问题。