塞沃尔-拉赛尔综合症: 遗传学基础和分子遗传学检测


Thomas Eggermann1, Matthias Begemann1, Gerhard Binder2, Sabrina Spengler1
1RWTH Aachen, Institute of Human Genetics, Aachen, Germany
摘要:父母源印记基因特异表达影响出生前生长的各个方面。因此可以预测许多目前已知的以生长障碍为临床特征的印记疾病(imprinting disorders,IDs)。赛尔沃-拉赛尔综合症(Silver-Russell syndrome,SRS)是一种重要的印记疾病,这是一种先天性疾病,以子宫内和出生后生长受限、相对巨头畸形、三角形面孔、不对称并在将来不典型为特征。但是临床谱宽阔,通常依主观临床诊断。仅50%的典型SRS特征的病人检测出遗传学和外成障碍,几乎1/10的病人携带有染色体7的母性单亲二体(UPD(7)mat),38%以上的病人表现为11p15内印记控制区域1的低甲基化,1%以上的病人表现为微观的染色体畸变。有趣的是,~7%的11p15低甲基化携带者,可检测到其它基因座的脱甲基化。临床上,这些病人与单纯11p15 低甲基化的病人没有区别,而UPD(7)mat的病人一般表现出轻度表型。但是,并不能够描述清晰的基因型-表型的关系。因此,我们提出诊断11p15低甲基化、UPD(7)mat和隐秘染色体不平衡的典型SRS表型病人的诊断程序。也可用于回忆有该疾病的轻度临床征兆的病人。
(Orphanet Journal of Rare Diseases 2010, 5:19)
回顾
最近20年来,越来越清楚低看到基因组印记与人类疾病密切相关。父母源印记基因的特异表达涉及到出生以前生长和行为的不同方面,目前已知的许多先天性印记疾病(imprinting disorders,IDs)以临床生长障碍为特征。虽然Angelman综合症、Prader-Willi综合症和Beckwith-Wiedemann综合症(BWS)是已经完全确认的IDs,但对于赛尔沃-拉塞尔综合症(Silver-Russell syndrome,SRS)的印记检测还是相对较新的结果。
SRS的主要特征为严重子宫内和出生后生长受限、相对巨头畸形、典型矮小和三角脸。这种疾病还伴有第五手指先天性侧弯和半发育不全(表1)的畸形特征。虽然最近提出了辅助诊断的临床打分系统,但诊断的准确性受到临床医生经验的影响。而且,成年SRS的临床症状不如儿童期初期那样清晰。
目前SRS的发生率尚未知,但由于特征范围宽大而存在漏诊的可能性。
SRS家族性病例以及细胞发生畸变的典型遗传学发现,证实了SRS病因中遗传学因素的影响。大部分SRS病例为散发的,但已经报告了家族性病例。Duncan et al.提出,大部分家族性病例以常染色体显性方式遗传,具有显著的家庭内可变性。在8个家庭也假设存在常染色体隐性遗传,但其中的6个家庭的临床病历存有疑问,但在仅有的2个家庭中就多次出现了11p15后成突变。
在下面,将讨论目前已知的SRS后成突变。我们要指出的是突变的次序并不反映重要性,而是鉴别的年代。
染色体畸变与SRS
几名SRS病人具有许多染色体受累的结构畸变,但只有染色体7,11和17符合严格SRS诊断标准。根据两名17q24-q25病人的平衡易位,曾经长时间讨论了该染色体区域在SRS病因中的重要作用。但是,两名病人17q断点的特性表明二者是不一样的。目前认为,所报告的17q上生长激素基因簇的杂合性缺失是一种致病的多态性。
然而,过去使用低微观分辨率妨碍了常规细胞发生学分析,因此,现在的阵列技术的分子学核型分析可鉴别出以前微观分析所不能的隐秘不平衡。在这期间,两项研究报告了携带<3Mb的小缺失/重复的SRS病人。关于遗传咨询,应当考虑病人父母的常规核型分析,以检测平衡的重排。
染色体7与SRS
在几名SRS病人,已经鉴别出的染色体7的细胞遗传学异常包括7p11.2p13重复和小标记染色体。该染色体成为病因的最初证据为,~10%的SRS病人鉴别出了染色体7的母性单亲二体(表1),几乎所有这些UPD(7)mat携带者的微卫星分型模式均一致性地具有三染色体自救机制。由于UPD(7)mat源自三染色体,所以可以假设三体性7镶嵌性(trisomy 7 mosaicism)是SRS的病因。这个假设被SRS病人完全偏斜的X失活频数的增加所确证,X失活是未在病人和/或胎盘检测出三体性7的生物学标志。不过,两项研究在SRS病人的白细胞和成纤维细胞中均未检测出三体性7细胞,可能是因为三体性7镶嵌性的致命性和所分析的细胞数量有限。
在UPD病例,隐性等位基因的纯合性是异常表型的又一病因,而且,确实是首先在生长延迟的囊性纤维化病人报告了UPD,这名病人因UPD(7)mat引起纯合子囊性纤维化跨膜调节基因突变。因此,隐性突变可能是这名UPD(7)mat病人SRS表型的病因,但是不存在常见的单亲二体型部分,这个结果排除了隐性基因是直接导致SRS的病因。
总之,UPD(7)mat携带者SRS表型最可能的解释是染色体7上印记基因失常表达。UPD(7)mat通常与生长延迟和SRS-样特征有关,而UPD(7)pat则无关。因此,假设:染色体7上的(i)父性基因表达的减少或(ii)母性基因的过表达引起SRS。
迄今为止,对染色体7编码因子的研究集中在了7p和7q染色体片段上面。对于7p11.2-p13候选区域,已经报告了SRS病人的重复,这个区域含有印记基因(生长因子受体结合蛋白10/GRB10)和几种涉及人类生长和发育的因子(IGFBP1; IGFBP3; PHKG1; EGFR; GHRHR)。在SRS,已经排除了这些基因的病理性突变,特别是,GBR10对生长具有重要作用,因此仍然是SRS的候选者。这个假设得到了最近发表的携带母亲遗传的dup(7)(p11.2p12)的家庭并未包括GRB10,也无SRS特征的支持。然而,尽管进行了广泛的筛查研究,但在SRS病人的这个编码区域即未检测出点突变,也未检测出GRB10的异常甲基化。
另一方面,也有SRS病因涉及到染色体7q31区域的证据:已经鉴别出有UPD(7q)片段的4名生长延迟病人,在7q31中有3个印记基因(MEST/PEG1; CPA4; COPG2)和2个非编码RNAs(MESTIT, CIT1/COPG2IT1),但筛查研究未检测出病理性变异体。此外,在SRS病人的MEST/PEG1基因座仍未检测出其它印记疾病所报告的单纯印记缺失。
也有报告,其它染色体UPDs引起的子宫内生长延迟,通常与人类妊娠期胎盘镶嵌性有关,几项报告在SRS病人使用微卫星分型研究了其它染色体,但并未观察到其它的UPDs。
染色体11p15与SRS
最大的SRS的分子遗传学亚组是11p15染色体区域后成突变和突变的病人。这个区域涉及SRS病因的最早的证据是在生长延迟病人鉴别出了母性11p15重复,6名中的4名病人,除了子宫内生长受限和出生后生长延迟外,表现出SRS特征。有趣的是,与其相对的障碍-父性11p15重复与BWS有关。在BWS病人可检测出许多遗传与外成的变化,但50%以上为11p15的异常甲基化。因此,才在SRS病人搜索11p15后成突变,并确实在38~63%的病人发现调节H19和IGF2表达的11p15内端粒ICR1低甲基化(表2)。
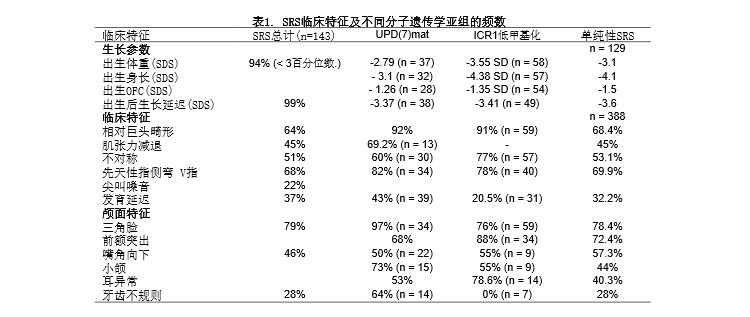
11p15印记簇包括有许多印记基因,其表达受到2个不同印记控制区域(ICR1和ICR2)的调节,也分别称为H19 DMR(分化的甲基化区域)和KvDMR1(见图1),对于胎儿的生长至关重要。
端粒ICR1调节2个父母等位基因特异的染色质结构,导致H19和IGF2呈彼此相反的表达。在胚胎发育过程中,2基因在源自内胚层和中胚层的组织中表达,竞争相同的增强子。父性表达的IGF2参与胎儿的发育和生长。虽然H19是最早鉴别出的非编码转录物之一,但是它的功能仍然不清楚。敲除H19,去除全部RNA编码序列但保留启动子和周围转录单位并不影响IGF2的印记表达。这些结果说明,RNA本身不具功能,但是在哺乳类,H19是相对高度保守的基因(人和鼠之间有77%的一致性)的事实提示了密切的功能联系。最近的研究表明,H19功能是作为基本的小RNA前身物,在脊椎动物发育过程中参与特定mRNA转录后的下调。在H19下游2 kb分化的甲基化区域内,ICR1有7个CTCF靶位点(CTCF1- CTCF7),锌指结合因子CTCF结合母性未甲基化的ICR1拷贝,因此形成染色质边界。这种CTCF结合机制阻断了IGF2表达,促进母性11p15拷贝H19的转录。
着丝粒ICR2调节CDKN1C, KCNQ1(钾通路KQT家族成员1)和更多基因彼此相反的表达,并只在母性等位基因上甲基化。在40%的家族性BWS和5~10%的散发病例,父性的突变抑制CDKN1C基因(表1)。该基因编码细胞周期蛋白依赖性激酶抑制因子(p57KIP2),是p21CIP2Cdk抑制因子家族的一部分。两名BWS病人CDKN1C胚层突变的功能分析证明了细胞周期抑制的降低。11p15内另一个非编码RNA的基因- KCNQ1OT1 (LIT1)位于KCNQ1基因的内含子9。KCNQ1OT1由父性等位基因表达,可能抑制CDKN1C基因功能的实现。母性ICR2等位基因的甲基化降低(LOM)与KCNQ1OT1的表达有关。在BWS,ICR2后成突变(ICR2低甲基化)以及CDKN1C点突变引起的一个重要生理学变化是CDKN1C的表达减少。
SRS的11p15后成突变主要是端粒ICR1低甲基化(表1)。与此相反,BWS最常见的变化是~50%的病人着丝粒ICR2低甲基化,而仅在2~7%的BWS病人诊断为ICR1超甲基化。临床上,大多ICR1低甲基化携带者符合SRS临床标准,但在仅有生长延迟和不对称的病人也已经诊断出后成突变。可是,在单纯出生前和出生后生长受限的病人尚未监测到这种障碍。
最近鉴别的SRS染色体重复的病人仅限于ICR2,提示了11p15上的两个ICRs都是这种疾病的病因,如同BWS。该结果和由BWS病人获得的进一步的数据,以及在小鼠的研究提示,ICR1和ICR2相互作用。
11p15低甲基化的孕后起因与SRS的多基因座异常甲基化
几乎所有SRS病人11p15后成突变的镶嵌分布都可能贡献于孕后错误。临床上,这种嵌合体反映为偏侧发育不全,大部分病人均存在这种临床表现。
双生子研究数据与后成突变的嵌合体分布相一致。在SRS,观察到了4对不一致和仅有的1对一致的单卵双生对。这种不明确的结果与Gicquel et al.的数据相一致。他们报告了血液细胞ICR1后成突变携带者双生子对的不一致,但受累的双生子皮肤成纤维细胞中存在LOM。对于ICR2基因座也有类似的观察,不一致的BWS双生子对的淋巴细胞有相同外成缺陷,但在成纤维细胞或口腔粘膜中有不同的甲基化模式。最终,ICR1或ICR2后成突变的孕后起因解释了SRS或BWS双生子对不一致的高发生率。
确定印记标记中的合子后缺陷的进一步证据来源于短暂的新生儿糖尿病(TNDM)病人,以及除了甲基化缺陷外有更多母性(和父性)印记基因座LOM的SRS和BWS病人的观察。在TNDM情况下,又有LOMs表现的病人表型与TNDM和仅在6q24有LOM病人稍有不同,可能是由于其它印记基因座甲基化改变所致。根据这些研究结果,Mackay et al.提出了母性低甲基化综合症的存在。最近,也报告了血液淋巴细胞多基因座低甲基化的BWS和SRS病人。这些病人白细胞中的父性和母性印记基因座都受累。在不一致的BWS单卵双生子,Bliek et al.在双生子对双方白细胞中观察到了类似的一个或多个基因座印记异常,但在颊粘膜DNA中,只在受累的双生子中可检测到后成突变。在所有的研究中,有多重低甲基化基因座的BWS或SRS病人与携带单纯11p15后成突变病人之间的表型差异不明显。
总结不同疾病的数据,异常甲基化镶嵌体提示受孕后出现了外成错误,影响了印记基因座甲基化信号的保持。原生殖细胞中的甲基化模式被大部消除,特定性别的甲基化模式在成熟的男女生殖细胞中重新建立。这个过程的关键调节因素是DNA甲基转移酶和甲基结合域蛋白,但是这些机制仍然在很大程度上未阐明。Howell et al.曾提出胚胎早期甲基化模式动力学复杂性的观点以及建立和保持基因组甲基化模式的机制:DNA甲基转移酶-1(Dnmt1)缺乏小鼠,在卵母细胞中正常建立基因组印记,但意想不到的是合子后甲基化的丧失。要维持印记基因座的甲基化可能需要Dnmt1,并且只在胚胎早期中的单一S时相中。在Dnmt3L-/-小鼠后代首次证实了规律印记的确立:当缺乏DNA甲基转移酶3L(Dnmt3L)时,其它因子标记了各个特异甲基化区域(DMRs),但对于所有基因座的适当印记模式,所有涉及因子的联合是必需的。
SRS和SRS-样特征病人的诊断程序
使用11p15内ICR1低甲基化和UPD(7)mat的鉴别,可对~50%的病人进行SRS临床诊断的分子学确证(图2)。
目前所有已知UPD(7)mat的病人是染色体不分离事件的结果,在这些情况下不增加家庭中的再现风险。其间,报告了几名染色体7长臂UPD的病人。因此,检测染色体7短臂和长臂已知印记基因座UPD(7)mat是有意义的。我们提议,使用甲基化特异的PCR方法检测7p和7q基因座,因为这样做不仅为诊断而检测病人的UPD(7)mat,而且也可以检测目前未知的染色体7上的单纯印记缺陷。如果获取阳性检测结果,则需要微卫星分型证实UPD(7)mat,排除前述的单纯性印记缺陷和缺失。
在出生前检测UPD的情况下,特异甲基化检测可能受到甲基化是否完备的不确定性的妨碍,因此微卫星分型是首选方法。
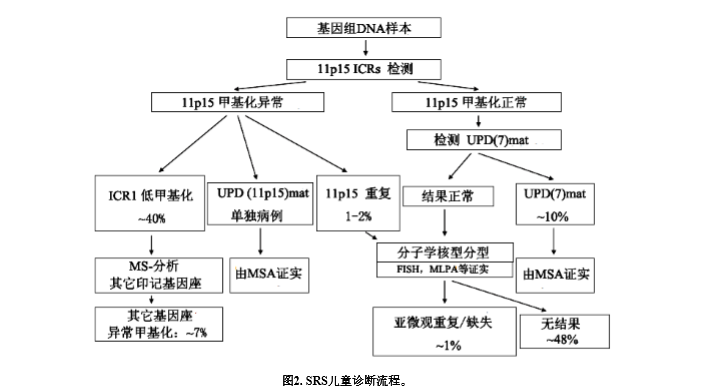
诊断程序包括MS-MLPA分析11p15基因座和染色体两臂基因座的UPD(7)mat。MSA: 微卫星分析; MS分析: 特异甲基化检测)
大多数SRS病人表现出11p15内ICR1低甲基化。已经报告几种检测方法可用于11p15基因座的甲基化分析,甲基化特异的多重探针依赖性分析方法(MS-MLPA)的优点是在单管中可以检测11p15内不同基因座的拷贝数变异和异常甲基化,因此可以鉴别11p15内ICRs甲基化缺陷和重复,以及该区域的UPDs。但是MS-MLPA以及报告的其它检测方法也具有局限性,例如影响探针杂交或重亚硫酸盐转换的序列变异体。此外,我们不要忘记,常规诊断依据于淋巴细胞,而且几乎所有ICR1低甲基化的SRS病人为嵌合体。因此,我们假设,少数病人逃逸了分子学诊断,因为他们的镶嵌性影响了血液细胞以外的组织。因此,在临床强烈怀疑SRS而又排除了主要的后成障碍的情况下,我们建议分析另一种细胞系统,例如口腔上皮细胞。尽管异常甲基化的MS-MLPA模式是明确的,并且不需要再次检测来证实,但重复/缺失和UPD要使用11p15标记微卫星分型或qPCR来进行核实。当前,对于11p15印记区域特异甲基化检测方法是否适用于出生前尚难以估价,因为胚胎中特定基因座甲基化开始时间不确定。
最近,在~7%的病人证实了除ICR1之外的其它基因座多种低甲基化。单纯ICR1低甲基化病人与多印记缺陷病人之间的临床差异普遍不明显。为了研究的目的,可检测ICR1脱甲基化病人的其它印记基因座,在排除了11p15后成突变和UPD(7)mat后,分子学核型分析可有助于鉴别亚微观的不平衡。SRS染色体不平衡的出现频数的确尚不了解,但根据对这一问题的2项研究,我们估价~1%的SRS病人存在这种畸变。
通过概括常规诊断SRS病例的分子遗传学数据,我们证明了11p15后成突变和UPD(7)mat携带者并不总是表现为明确的SRS表型,在“SRS-样”表型的病例也应当进行这两种畸变的遗传学检测,例如,仅有前额突出和三角脸或不对称临床特征的轻微子宫内生长受限和出生后生长延迟(>-2SD)病人。特别是,不要机械地将有“SRS-样”表型的非IUGR病人排除在分子学检测之外。
总之,鉴于SRS的临床诊断的主观性,SRS的分子学验证特别重要。关于遗传咨询,ICR1低甲基化或母性UPD7的鉴别由于重新发生而有低的再现风险。最初的临床特征提示母性UPD7携带者的表型一般较轻,而11p15后成突变携带者通常表现出典型的SRS特征。进一步的表型分析将有助于阐明Binder et al.所提出的SRS的分子学亚组是否对生长激素治疗有不同的反应。